SCREE
Last updated: 2023-06-03
Checks: 7 0
Knit directory: SCREE/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210907) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version dc5b2b2. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Untracked files:
Untracked: data/workflow.png
Untracked: img/
Untracked: output/workflow.png
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/SCREE.Rmd) and HTML (docs/SCREE.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 2c16336 | HailinWei98 | 2021-09-23 | Build site. |
| Rmd | a2a5b0a | HailinWei98 | 2021-09-23 | Publish the initial files for myproject |
| html | eeeebf3 | HailinWei98 | 2021-09-23 | Build site. |
| Rmd | 94450d1 | HailinWei98 | 2021-09-23 | Publish the initial files for myproject |
| html | 94450d1 | HailinWei98 | 2021-09-23 | Publish the initial files for myproject |
| html | 518a5ca | HailinWei98 | 2021-09-22 | Build site. |
| Rmd | ab04ccf | HailinWei98 | 2021-09-22 | Publish the initial files for myproject |
| html | 855bd74 | HailinWei98 | 2021-09-22 | Build site. |
| Rmd | e582e5c | HailinWei98 | 2021-09-22 | Publish the initial files for myproject |
SCREE(Single-cell CRISPR scREen data analyses and pErturbation modeliNg) is a comprehensive pipeline to visualize data quality and model perturbation effect of single-cell CRISPR screens RNA-seq/ATAC-seq datasets. SCREE has integrated three R functions: scMAGeCK_lr, Mixscape and plot function of cicero. These functions are used to model the regulatory score between perturbations and genes, estimate the perturbation efficiency of each perturbation and visualize enhancer potential targets, respectively.
Schema
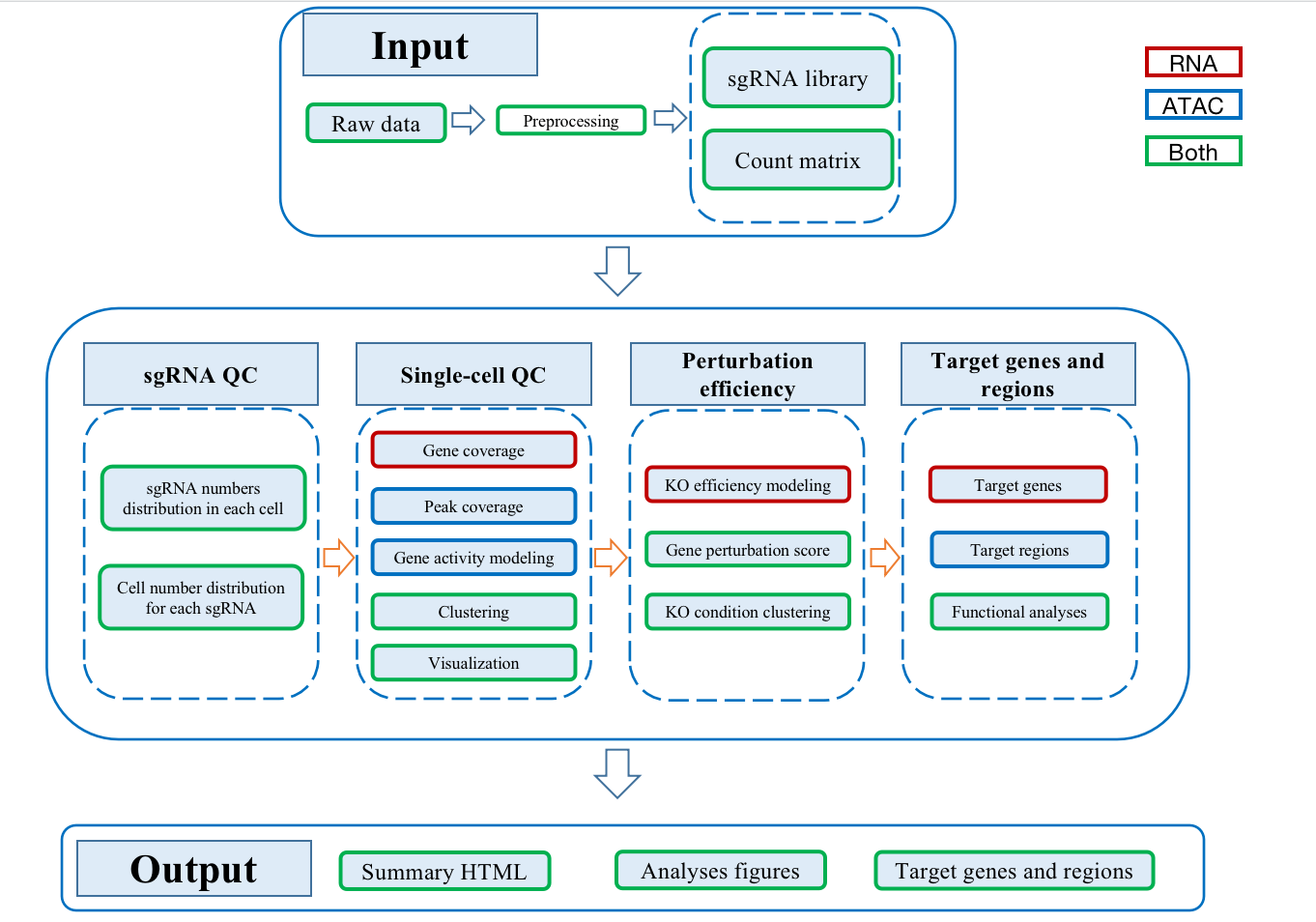
Usage
SCREE provide two major functions, preprocess and analysis. To get a full list of subcommands and descriptions:
usage: SCREE [-h] [-v] {preprocess,analysis} ...
| Subcommands | Description |
|---|---|
preprocess |
Reads alignment and quantification. |
analysis |
Performing all downstream analyses with one command. |
Preprocess
The usage of the preprocess function:
usage: SCREE preprocess [-h] [--datatype {RNA,ATAC}]
[--filetype {multi,single}] [--reference REFERENCE]
[--config CONFIG] [--mkref] [--input INPUT]
[--sample SAMPLE] [--output OUTPUT] [-m MEMORY]
[-c CORES] [--csv CSV] [--bin] [--binsize BINSIZE]
[-n PROCESS_N] [-l CHRLENGTH] [--para PARA]
| Options | Description |
|---|---|
--help/-h |
Show this help message and exit. |
--filetype |
Input file type, one of 'multi', 'single'. 'multi' indicates sgRNA sequence and mRNA/DNA sequence are not in the same files, 'single' indicates sgRNA sequence and mRNA sequence are in the same files. Notably, for scATAC-seq input, SCREE only support 'multi'. |
--reference |
Reference path. |
--config |
Config file to generate reference index. |
--mkref |
Generate reference index or not. |
--input |
File path of all the fastq files. |
--sample |
Sample prefix. |
--output |
Output folder to create. |
-m |
Maximum local memory to use. Unit: GiB. |
-c |
Maximum local cores to use. |
--csv |
Config file to process 'multi' file type. |
--bin |
Generate a bin-based matrix instead of a peak-based matrix. |
--binsize |
Size of the genome bins to use. Only available with the parameter '--bin'. Default is 5000. |
-n, --process_n |
Number of regions to load into memory at a time, per thread. Processing more regions at once can be faster but uses more memory. Only available with the parameter '--bin'. Default is 2000. |
-l, --chrLength |
Path to the file which stores the chromosome length of the reference genome, always named as 'chrNameLength.txt'. Only available with the parameter '--bin'. |
--para |
Other parameters passed into cellranger or cellranger-atac, should be in string format like '--parameter1 value1 --parameter2 value2'. |
Analysis
The usage of the analysis function:
usage: SCREE analysis [-h] [--type {RNA,ATAC,enhancer}]
[--replicate REPLICATE] [--mtx MTX] [--sg_lib SG_LIB]
[--sg_format {10X,dataframe,matrix,table}]
[--freq_cut FREQ_CUT] [--freq_percent FREQ_PERCENT]
[--unique] [--project PROJECT] [--species {Hs,Mm}]
[--fragments FRAGMENTS] [--prefix PREFIX]
[--label LABEL] [--feature_frac FEATURE_FRAC]
[--nFeature NFEATURE] [--nCount NCOUNT]
[--percent.mt PERCENT.MT] [--blank] [--NTC NTC]
[--sg.split SG.SPLIT] [--raster]
[--ref_version {v75,v86,v79}]
[--gene_type {Symbol,Ensembl}] [--pval_cut PVAL_CUT]
[--score_cut SCORE_CUT] [--article ARTICLE]
[--article_name ARTICLE_NAME] [--data DATA]
[--data_name DATA_NAME] [--padj_cut PADJ_CUT]
[--logFC_cut LOGFC_CUT]
| Options | Description |
|---|---|
--help/-h |
Show this help message and exit. |
--type |
Data type, one of 'RNA', 'ATAC', 'enhancer'. Default is 'RNA'. |
--replicate |
Directory to a txt file only contains the replicate information of each cell, in the same order of cells in the input matrix. If no replicate information, we will consider that all cells are from the same replicate, and this parameter will be set as 1. Default is 1. |
--mtx |
Directory to an rds file of the SeuratObject, with cell in columns and features in rows, or the directory to the folder of 10X-like outputs. |
--sg_lib |
Directory to a txt file of the sgRNA information. This parameter can be ommited when the 'sg_format' is '10X'. |
--sg_format |
Format of the input sg_lib, one of '10X', 'dataframe', 'matrix', 'table'. '10X' means the sgRNA information is an sgRNA count matrix stored in the 10X-like outputs; 'dataframe' means a table with 3 columns: cell(cell barcode), barcode(sgRNA name), freqUMI counts in each cell for each sgRNA; 'matrix' means a sgRNA count matrix with cells in columns and sgRNAs in rows; 'table' means a table with 3 columns: cell(cell barcode), barcode(sgRNA name), gene(sgRNA targeted gene). Default is '10X'. |
--freq_cut |
Cutoff of sgRNA count numbers. For each cell, only sgRNA with counts more than freq_cut will be retained. Default is 20. |
--freq_percent |
Cutoff of sgRNA frequency. For each cell, only sgRNA with frequency more than freq_percent will be retained. Default is 0.8. |
--unique |
Only retain cells with the top target that passed the freq_percent, which means each cell will be assigned with a unique sgRNA. |
--project |
Project name. Default is 'perturb'. |
--species |
Species source of input data, one of 'Hs', 'Mm'. Defaule is 'Hs'. |
--fragments |
Directory to the fragments file. Only required for scATAC-seq based input. |
--prefix |
Path to the results generated by SCREE. Default is current directory. |
--label |
The prefix label of previous output file. Notably, there needs a separator between default file names and the label, so label would be better to be like 'label_'. Default is ''. |
--feature_frac |
A paramter for filtering low expressed genes or low accessible peaks. By default, only genes/peaks that have counts in at least that fractions of cells are kept. Default is 0.01. |
--nFeature |
Minimal count numbers in each cell. Default is 200. |
--nCount |
Minimal detected feature numbers in each cell. Default is 1000. |
--percent.mt |
Maximum mitochondrial gene percentage of each cell. This parameter can also be used for scATAC-seq based data to represent the minimum fraction of reads in peaks(FRiP). Default is 10(means 10%%). |
--blank |
Use blank control as negative control. With this parameter, cells assigned with no sgRNA will be removed. |
--NTC |
The name of negative controls. Default is 'NTC'. |
--sg.split |
String to split sgRNA name. Default is '_sgRNA', which means sgRNA named in the format like 'gene_sgRNA1'. |
--raster |
Convert points to raster format, will be useful to reduce the storage cost of the output figure or pdf. |
--ref_version |
Version of the reference genome(Ensembl), one of 'v75'(hg19/mm9), 'v79'(hg38/mm10), 'v86'(hg38). Default is 'v75'. |
--gene_type |
Type of gene names, can be one of 'Symbol', 'Ensembl'. Default is 'Symbol'. |
--pval_cut |
P-value cutoff of improved_scmageck_lr results. Default is 0.05. |
--score_cut |
Score cutoff of improved_scmageck_lr results. Default is 0.2. |
--article |
The link to the article of the dataset. |
--article_name |
Name of the article. |
--data |
Link to the data source. |
--data_name |
Name of the data, usually the GSE accession number. |
--padj_cut |
Maximum adjust p_value. Default is 0.05. |
--logFC_cut |
Limit testing to genes which show, on average, at least X-fold difference (log-scale) between the two groups of cells. Default is 0.25. |
10X config file
The latest version of cellranger can be used to align single-cell CRISPR screens RNA-seq data, which need a config file and a sgRNA reference file as input. More details of the config file can be found in Cellranger multi. Here is the example of config file:
[gene-expression]
reference,/path/to/references/refdata-gex-GRCh38-2020-A
expect-cells,5000
[feature]
reference,/path/to/feature_refs/SC3P_CellPlex_Set_A_millipore_pool_v2_jul_2020.csv
[libraries]
fastq_id,fastqs,lanes,physical_library_id,feature_types,subsample_rate
SC3_v3_NextGem_DI_CRISPR_A549_5K_gex,/path/to/fastqs/SC3_v3_NextGem_DI_CRISPR_A549_5K/SC3_v3_NextGem_DI_CRISPR_A549_5K_gex,any,CRISPR_A549_5K_gex,gene expression,
SC3_v3_NextGem_DI_CRISPR_A549_5K_crispr,/path/to/fastqs/SC3_v3_NextGem_DI_CRISPR_A549_5K/SC3_v3_NextGem_DI_CRISPR_A549_5K_crispr,any,CRISPR_A549_5K_crispr,Crispr Guide Capture,
10X sgRNA reference in csv format
sgRNA reference file is used to do sgRNA alignment. Besides sgRNA names and corresponding target genes, sgRNA reference need the sequence of sgRNA constant region, the sequence next to the spacer sequence in fastq file. Here is the example of sgRNA reference file:
id,name,read,pattern,sequence,feature_type,target_gene_id,target_gene_name
Non-Targeting-5,Non-Targeting-5,R2,(BC)GTTTAAGAGCTAAGCTGGAA,ACTCGAAATCACCTATGGTA,CRISPR Guide Capture,Non-Targeting,Non-Targeting
Non-Targeting-7,Non-Targeting-7,R2,(BC)GTTTAAGAGCTAAGCTGGAA,TTATGTGAGCACGCCATTAC,CRISPR Guide Capture,Non-Targeting,Non-Targeting
Non-Targeting-8,Non-Targeting-8,R2,(BC)GTTTAAGAGCTAAGCTGGAA,CGACGGTAATGCACCTACTA,CRISPR Guide Capture,Non-Targeting,Non-Targeting
APH1A-1,APH1A-1,R2,(BC)GTTTAAGAGCTAAGCTGGAA,GGCAACGCGACCCCACGAG,CRISPR Guide Capture,ENSG00000117362,APH1A
APH1A-2,APH1A-2,R2,(BC)GTTTAAGAGCTAAGCTGGAA,ATGTCACCCCCAGACCCCG,CRISPR Guide Capture,ENSG00000117362,APH1A
CDKN3-1,CDKN3-1,R2,(BC)GTTTAAGAGCTAAGCTGGAA,TGCAGCGCCGGCGACTCAC,CRISPR Guide Capture,ENSG00000100526,CDKN3
CDKN3-2,CDKN3-2,R2,(BC)GTTTAAGAGCTAAGCTGGAA,CGGGGCACCGGTGAGTCGC,CRISPR Guide Capture,ENSG00000100526,CDKN3
EZR-1,EZR-1,R2,(BC)GTTTAAGAGCTAAGCTGGAA,CACTCGGCGGACGCAAGGG,CRISPR Guide Capture,ENSG00000092820,EZR
EZR-2,EZR-2,R2,(BC)GTTTAAGAGCTAAGCTGGAA,GCGCACTCGGCGGACGCAA,CRISPR Guide Capture,ENSG00000092820,EZR
GRB2-1,GRB2-1,R2,(BC)GTTTAAGAGCTAAGCTGGAA,TGCTGCTTCGGCGACCGGG,CRISPR Guide Capture,ENSG00000177885,GRB2
GRB2-2,GRB2-2,R2,(BC)GTTTAAGAGCTAAGCTGGAA,TTCTCGCGGGACACCGACG,CRISPR Guide Capture,ENSG00000177885,GRB2
GSK3A-1,GSK3A-1,R2,(BC)GTTTAAGAGCTAAGCTGGAA,AGCCCAAGCCAGAGCGGCG,CRISPR Guide Capture,ENSG00000105723,GSK3A
GSK3A-2,GSK3A-2,R2,(BC)GTTTAAGAGCTAAGCTGGAA,GAGCGGCGCGGCCTGGAAG,CRISPR Guide Capture,ENSG00000105723,GSK3A
HRAS-1,HRAS-1,R2,(BC)GTTTAAGAGCTAAGCTGGAA,ACCCGAGCCGCACCCGCCG,CRISPR Guide Capture,ENSG00000174775,HRAS
HRAS-2,HRAS-2,R2,(BC)GTTTAAGAGCTAAGCTGGAA,GCACGGGCGGCGGAGACTC,CRISPR Guide Capture,ENSG00000174775,HRAS
JUN-1,JUN-1,R2,(BC)GTTTAAGAGCTAAGCTGGAA,AGCAGGGCTCTCCTCCCGG,CRISPR Guide Capture,ENSG00000177606,JUN
JUN-2,JUN-2,R2,(BC)GTTTAAGAGCTAAGCTGGAA,TGTGGCTGAAGCAGCGAGG,CRISPR Guide Capture,ENSG00000177606,JUN
10X fastq filename
As SCREE alignment part is based on cellranger, names of input fastq file must in the same format:
SC3_v3_NextGem_DI_CRISPR_A549_5K_gex_S5_L001_I1_001.fastq.gz
sessionInfo()R version 4.0.2 (2020-06-22)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Catalina 10.15.7
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRblas.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] workflowr_1.6.2
loaded via a namespace (and not attached):
[1] Rcpp_1.0.7 whisker_0.4 knitr_1.33 magrittr_2.0.1
[5] R6_2.5.0 rlang_0.4.11 fansi_0.5.0 stringr_1.4.0
[9] tools_4.0.2 xfun_0.25 utf8_1.2.2 git2r_0.28.0
[13] jquerylib_0.1.4 htmltools_0.5.1.1 ellipsis_0.3.2 rprojroot_2.0.2
[17] yaml_2.2.1 digest_0.6.27 tibble_3.1.3 lifecycle_1.0.0
[21] crayon_1.4.1 later_1.2.0 sass_0.4.0 vctrs_0.3.8
[25] promises_1.2.0.1 fs_1.5.0 glue_1.4.2 evaluate_0.14
[29] rmarkdown_2.10 stringi_1.7.3 bslib_0.2.5.1 compiler_4.0.2
[33] pillar_1.6.2 jsonlite_1.7.2 httpuv_1.6.1 pkgconfig_2.0.3